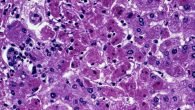
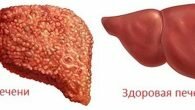
Гепаторентикулярная дегенерация, она же — болезнь Вильсона-Коновалова возникает при патологиях обменных функций меди. При отсутствии синтеза медь откладывается в органах, больше всего в печени и мозге. Это нарушает функционирование такого органа, постепенно вызывая развитие патологии. Возникает редко — по статистике, фиксируется 30 случаев на 1 миллион человек. Названа в честь докторов, которые впервые диагностировали эту болезнь.

Общие сведения
Расстройство такого вида метаболизма имеет врожденную форму. Спровоцирована патология нарушением синтеза белка, который переносит минерал. Вызывает заболевание ген АТР7 В, что находится в 13 хромосоме. Нарушается природный баланс наличия меди в организме: блокируется функция выделения элемента вместе с желчью и таким образом повышается ее уровень. Далее происходит обширная интоксикация организма. Поражается печень, почки и другие органы. Как следствие, вызывает цирроз печени, что присутствует во всех случаях этой болезни.
Вернуться к оглавлениюФормы болезни
- Брюшная — сильно поражается печень, возможен летальный исход. Чаще этой формой болеют дети. Длительность болезни от 2−3 месяцев до 5 лет.
- Ранняя — протекает быстро, возникает с детского возраста. Нарушается мышечный тонус, движения становятся медленными и скудными. Возможно нарушение интеллектуального развития. Длится до 3 лет, обычно имеет летальный исход.
- Дрожательно-ригидная — самая распространенная форма. Начинается в юности, развивается медленно с возможными этапами ремиссии. Во время резкого рецидива отмечается ригидность больного, сильный тремор, мышцы напряжены. Продолжается до 6 лет.
- Дрожательная — возникает во взрослом возрасте, как правило, до 30 лет. Развивается медленно — до 20 лет. Характеризуется сильной дрожью, замедлением речи, изменениями в психическом состоянии, возможно частое возникновение аффективного состояния. Могут наблюдаться эпилептические припадки.
- Экстрапирамидно-корковая — самая редкая форма. Характеризуется внезапно развивающимися общемозговым и очаговым неврологическими симптомами, нарушением мозговой функции пирамидной системы, проявляются парезы (частичное отсутствие или ослабление движений). Постепенно усиливается психическое расстройство, начинаются эпилептические припадки. Как правило, имеет летальный исход через 7−8 лет.

Причины развития
Формирование синдрома Вильсона-Коновалова предполагает 100% наследственность и только тогда, когда отец и мать являются носителями данного генного нарушения. Первоначальным источником болезни выступает патологическое изменение гена АТР7 В. Обнаружено больше 75 видов нарушения. Наибольшую опасность представляют те мутации, что полностью разрушают ген. Это ускоряет протекание болезни и приводит к тяжким формам заболевания.
Вернуться к оглавлениюЧто происходит?
Чем позже начинается развитие синдрома Вильсона-Коновалова, тем медленнее он протекает. Обычно отмечается здоровое поглощение меди в кишечном тракте, баланс нарушается отсутствием или ухудшением высвобождения меди с желчью. Это вызывает накопление микроэлемента в органах. Появляется дефицит или полное отсутствие протеина типа Р, что отвечает за транспортировку меди и ее высвобождения из организма. Медь не включается в церулоплазмин, что должен присутствовать в крови, что вызывает его дефицит.

Повышенный уровень меди вызывает ее первичное накопление в печени, поэтому заболевание чаще проявляется с симптомами патологии печени (в 49% примеров). Обычно нарушения проявляются в возрасте 7−11 лет, даже если уровень аминотрансфераз повышен с рождения. Насыщение печени медью не выражается никакими симптомами, далее она откладывается в остальных органах — почках, сердце, в центральной нервной системе, в головном мозге. Уровень меди может увеличиться в более чем 50 раз. Тогда происходят психические нарушения на нейронном уровне, что возникают в 20−30 лет.
В случаях, когда медь поступает в кровь большим количеством, она крепится на эритроцитах и образует белковые соединения. Со временем это развивает гемолитическую анемию. Если же медь откладывается на роговичной мембране, это происходит параллельно с проявлениями симптомов нейронных нарушений.
Вернуться к оглавлениюСимптомы болезни Вильсона – Коновалова
Симптомы могут проявляться по-разному, это зависит от картины развития заболевания и ее формы. В случае поражения печени на начальных стадиях отмечается появление жировой неспецифической дистрофии, изменяются гепатоциты и возникает перипортальный фиброз. По истечении времени начинается прогрессирование хронического гепатита, проявляется желтуха, повышается наличие аминотрансферозы и возникает гипергаммаглобулинемия.
Если болезнь Вильсона имеет дальнейшее развитие, возникает цирроз печени, который выражается недостаточностью печеночных клеток и стойким нарушением циркуляции в портальных сосудах. Редчайшим симптомом отмечают фульминантную недостаточность печени. Она характерна для молодых пациентов и подростков. Основным признаком такой недостаточности будет высокая активность трансаминаз с преобладающей активностью аспартатаминотрансферазы, понижение уровня альбумина и высокое количество меди и билирубина.

Визуально наличие излишней меди можно определить эффектом «кошачьего глаза» — глазная роговица приобретает яркий окрас с зелено-желтым оттенком. Медики называют этот синдром кольцом Кайзера-Флейшера. Внезапное проявление заболевания характерно возникновением желтухи, ощущается общая слабость, резко поднимается температура, пропадает аппетит. Мгновенно развивается печеночная недостаточность. В подавляющем большинстве случаев болезнь имеет летальный исход. Если заболевание протекает хронически, проявления могут отсутствовать вплоть до 5−6 лет. Часто заболевание начинается из поражения печеночной ткани, так как именно в ней начинает изначально откладываться медь.
Вернуться к оглавлениюНарушения печени и неврологии обнаруживаются одновременно, и если не начать своевременное лечение, существует риск развития этих двух форм заболевания. Повышенный уровень меди может вызывать развитие таких болезней, как сахарный диабет, болезни сосудов, атеросклероз, синдром Фанкони.
Диагностика
Диагностику проводит квалифицированный специалист, устанавливая сразу изначальное развитие и течение болезни со слов пациента. В первую очередь выясняется, присутствовал ли такой диагноз у родителей и родственников. Проводят осмотр роговицы глаз, пальпацию печени. Выявление изменения цвета роговицы происходит в 80% случаев без вспомогательных средств. Далее проводят лабораторные анализы:
- Общий анализ крови делают для диагностики осложнения заболевания в виде анемии, так как с помощью этого анализа нельзя диагностировать болезнь. Биохимический анализ крови при наличии болезни покажет значительное снижение церулоплазмина (ниже 20 миллиграмм на децилитр).
- Обнаруживается увеличение АЛАТ и АСАТ. Точность такого анализа — 90%. Определяется уровень меди. Повышенным он считается, если количество микроэлемента выше 1500мг/л.
- Проводится анализ мочи на наличие меди. При положительном результате она присутствует в размере 100−1000 мкг в сутки.
- Биопсия печени — анализ печеночной ткани на количественное содержание меди.
- Иногда используется метод под названием «меченая медь» — когда в организм внедряется специально помеченная медь и наблюдают ее транспортировку и накопление.
- Генетическое исследование — используются маркеры ДНК, которые дают возможность поставить практически 100%-й диагноз. Главным минусом такой процедуры является огромное разнообразие генных мутаций, которые имеет болезнь Вильсона.
Лечение
Положительный результат может показать только проведение лечения под наблюдением врача-гастроэнтеролога или терапевта. Главной целью является выведение излишков меди из организма, строгое контролирование поступления в организм меди с пищей. Лечение можно условно разделить на 3 вида:

- Медикаментозное лечение подразумевает использование препаратов, которые способствуют выведению меди из организма больного. Используется постоянно на протяжении всей жизни. Главным веществом, обладающим лечебным эффектом, является пеницилламин. Он приводит в норму процесс переработки меди и ее успешного метаболизма. При непрерывном правильном лечении в среднем через 4 года у большинства пациентов (более 75%) исчезают симптомы заболевания.
- Немедикаментозное лечение построено на ограничении поступления меди в организм. Для этого применяется так называемая диета № 5 — запрещается принимать жирную, жареную и острую еду, а также ту еду, в которой содержится много меди (шоколад, орехи, бобовые), морепродукты, сухофрукты. Запрещается употреблять алкоголь, гепатотоксические препараты (нестероидные противовоспалительные медикаменты, аминогликозиды и др.). Можно пить только дистиллированную воду, рекомендуется отказаться от употребления соли.
- Хирургическое лечение назначается тем, у кого развилась портальная гипертензия. Проводится трансюгулярное шунтирование печени. Для излечения вен желудочно-кишечного тракта или асцита также пользуются методами хирургии. При фулминантной недостаточности печени или прогрессирующем циррозе показана ортотопическая трансплантация этого органа.
Вернуться к оглавлениюЕсли больной с абсолютной точностью проходит лечебный курс, у 80% случаев прогноз на будущее положительный. Пациент избавляется от проявлений болезни и в дальнейшем может заниматься трудовой деятельностью. Неблагоприятный прогноз может быть лишь в случае диагноза печеночной недостаточности.
Возможные осложнения

Последствия, которые может спровоцированы болезнью Вильсона-Коновалова:
- Печеночная недостаточность либо цирроз печени — образование на органе рубцовой ткани. Во втором варианте — одиночное или множественное нарушение функций органа, вызванные разрушением гепатоцитов (клеток печени).
- Асцит — жидкость скапливается в брюшине и как следствие может развиться воспаление живота.
- Повреждение вен в пищеводе — разрушение целостности стенок из-за постоянного давления на них изнутри и возможное появление кровотечения как усугубление патологии. Этому может сопутствовать рвота с кровью, кал черного цвета, спад артериального давления и учащение сердечного ритма.
- Печеночная энцефалопатия — нарушения сознания, нервное расстройство, что проявляется из-за нарушения или отсутствия необходимых функций печени.
- Опухоль на печени — гепатоцеллюлярная карцинома, что приводит к отсутствию фильтрации и следствием будет обширная интоксикация крови.
- Патология кишечника либо желудка из-за нарушения циркуляции крови.
- Патологическое протекание месячных — отсутствие регулярного цикла, очень обильный или скудный менструальный цикл, его полное отсутствие.
- Возможно бесплодие как результат поражения эндокринной системы.
Профилактика
Определенной профилактики, направленной против развития болезни, не существует. Это объясняется врожденной причиной болезни, которая зарождается еще в утробе и передается «по наследству» на генном уровне. Профилактические действия могут быть направлены на предупреждение развития каких-либо синдромов и осложнений болезни.
Для детей необходимы регулярные осмотры педиатра (не менее 3−5 раз за год до 7-летнего возраста, и до 3 раз в год для детей до 15 лет). Если обнаруживается малейшее развитие обострений, заметен даже незначительный синдром, осмотры необходимо делать минимум в 2 раза чаще. Строгий контроль принятия пищи, минимизация продуктов, содержащих медь, запрещено употребление запрещенных врачом медикаментов. Необходимо вести здоровый активный образ жизни, отказаться от курения и алкоголя. Желательно принимать поливитаминные добавки, подвергать профилактике воспаления печени. Нужно следить за здоровьем желудочно-кишечного тракта.
Избежать тяжелых последствий поможет своевременная постановка диагноза. Болезнь Коновалова должна быть обнаружена еще в пренатальном периоде (до рождения плода). Такая диагностика проводится врачом-генетиком. Это называется первичной профилактикой. Вторичной будет назначение курса лечения для предотвращения проявлений болезни, для этого проводятся необходимые анализы. По истечении 4−8 лет (длительность лечения носит индивидуальный характер) проводится повторное проведение обследования. Пациент постоянно должен наблюдаться у лечащего врача и стоять на учете в больнице.

 (Уже оценили: 10)
(Уже оценили: 10)